
What regulatory challenges do Software as a Medical Device Platforms face?
Many healthcare software products are described as “platforms”. This article explores how that concept applies to Software as a Medical Devices (SaMDs), including AI as a Medical Devices (AIaMDs) – focusing on regulatory considerations and the underlying technical architecture. This article builds on our previous article about platforms as marketplaces.
First, what is a platform?
Simply put, a platform is a software product or service that allows both its creators and other companies to deploy complementary products. In this context, those products are typically Medical Device Software (MDSW), including AIaMD applications.
There are a couple of definitions that are useful to describe platforms. The US Department of Commerce’s National Institute of Science and Technology (NIST) Definition of Cloud Computing publication – and recently leveraged in the US FDA’s Computer Software Assurance for Production and Quality System Software) – help to frame this. Here, I paraphrase to use the terms Platform manufacturer and MDSW manufacturer:
Software as a Service (SaaS): The capability provided to the MDSW manufacturer is to use the Platform manufacturer’s applications running on a cloud infrastructure. The applications are accessible from various client devices through either a thin client interface, such as a web browser (e.g., web-based email), or a program interface. The MDSW manufacturer does not manage or control the underlying cloud infrastructure, including network, servers, operating systems, storage, or even individual application capabilities, with the possible exception of limited user-specific application configuration settings.
Platform as a Service (PaaS): The capability provided to the MDSW manufacturer is to deploy MDSW manufacturer-created or acquired applications onto the cloud infrastructure, using programming languages, libraries, services, and tools supported by the Platform manufacturer. The MDSW manufacturer does not manage or control the underlying cloud infrastructure including network, servers, operating systems, or storage, but has control over the deployed applications and possibly configuration settings for the application-hosting environment.
A platform and its MDSW applications can generally be described using a three-tier architecture:
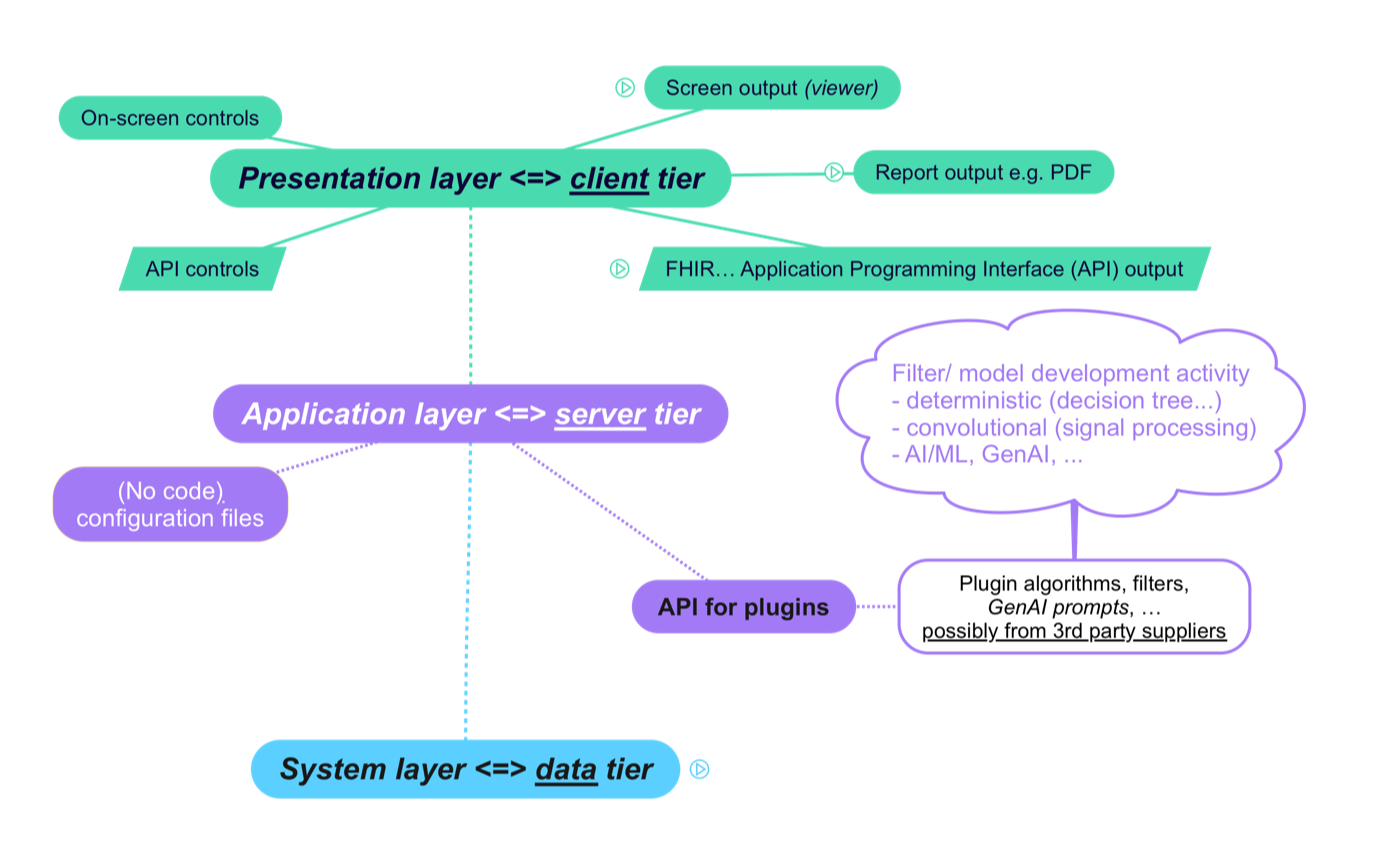
The presentation layer is what the end user interacts with, whether a human user interacting via a User Interface (UI) or another machine interacting via an Application Programming Interface (API). The presentation layer may be implemented on client hardware, such as a desktop or laptop computer, and/or a mobile device or tablet; it may also be implemented in a web browser.
The application layer is where the core MDSW functionality resides, either on a cloud or physical server or within the same device as the presentation layer.
The key point here is treating the various MDSW functionalities as plugins, with an API that standardises the way that these plugins interact with the Platform.The system layer is what facilities common services for the application and presentation layers, such as user authentication, license (revenue) management for use of the Platform and its apps, connections to data sources, software upgrade and rollback (e.g. for security patches and functionality upgrades, and the ability to revert to previous versions), etc. as illustrated in the following diagram:
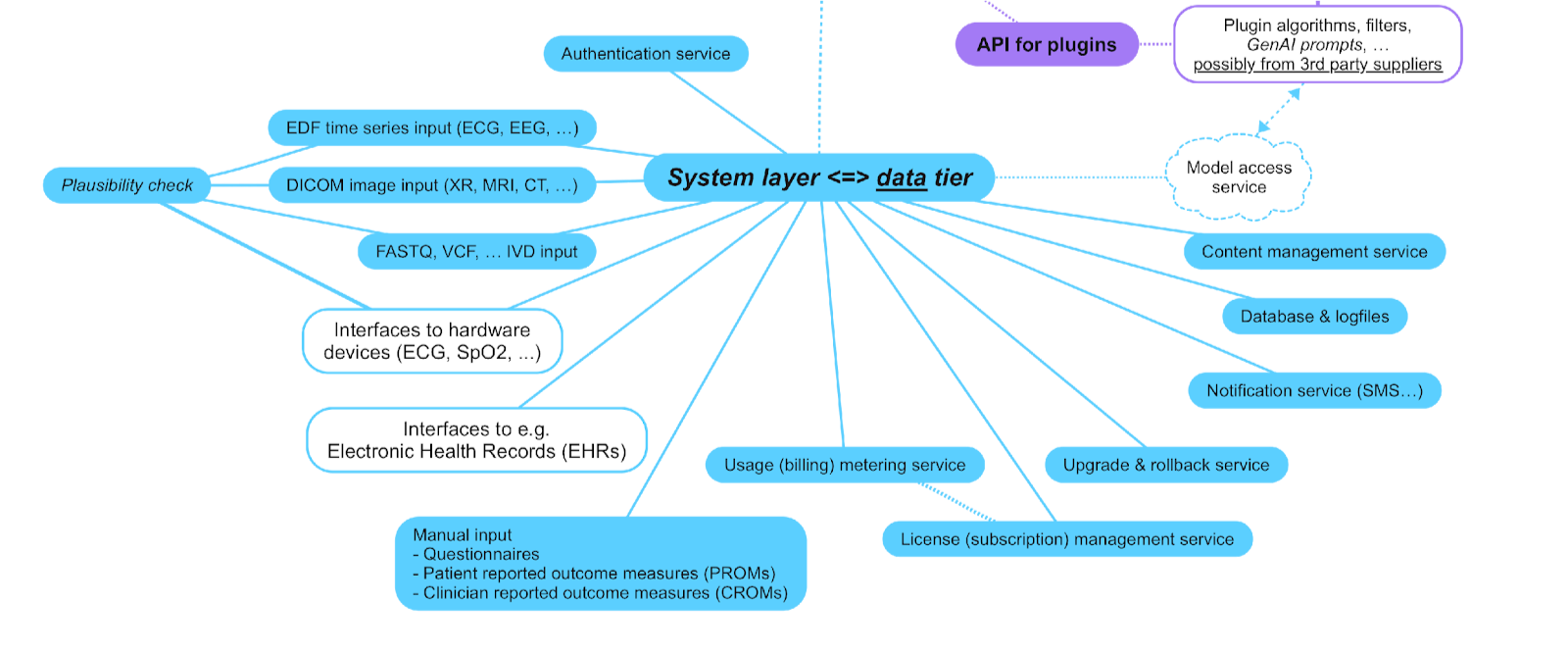
This structure is common in various medical device scenarios, including:
Radiology and AI/ML tools: Platforms like Picture Archiving and Communications Systems (PACS) or Medical Image Management and Processing System (MIMPS) underpin Computer-Aided Triage (CADt), Computer-Aided Detection (CADe), or Computer-Aided Diagnosis (CADx)..
Predictive and clinical documentation tools: Ambient Voice Technologies (AVT) – defined as AIaMD capable of capturing spoken clinical interactions and generating written documentation such as summaries or referral letters through Generative AI or Large Language Model (LLM) technologies – can be integrated within Electronic Health Record (EHR) systems. For secondary care scenarios, including the generation of discharge summaries, EHR-based platforms and/or Federated Data Platforms (FDPs) may serve as a similar foundation for these applications.
Agentic AI: Systems that proactively pursue defined goals – for example, generating discharge summaries or referral documentation from structured EHR data – rely on the same platform principles.
Digital Therapeutics (DTx) and Companion Diagnostics (CDx): Multiple therapeutic and diagnostic applications can be delivered through a single platform interface, sharing system resources and data connections to maintain consistency and traceability across products.
So, what are the regulatory challenges?
The first thing to consider is the qualification and classification of each MDSW app and of the Platform itself.
It should be clear that each MDSW app may in its own right be qualified as a SaMD, as per the regulatory jurisdiction into which it is intended to be deployed. Take clinical calculators as a case in point: the UK MHRA states that some clinical calculators are not medical devices and some are, based on a level of functionality:
Unlikely to be a medical device
“Calculators without a medical purpose” e.g. those performing statistical analysis of clinical data, not for the benefit of individual patients
“Simple scores”
“Simple calculations”
Likely to be a medical device
“Intermediate calculations"
“Complex calculations”
“Calculators with linked lookup tables” including calculations pulling data from fields in a patient’s electronic health record (EHR) rather than having the data entered manually by the user
Then comes the classification of each MDSW app:
-
Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
Death or an irreversible deterioration of a person's state of health, in which case it is in class III; or
A serious deterioration of a person's state of health or a surgical intervention, in which case it is classified as class IIb.
Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.
All other software is classified as class I.
-
Stand-alone software is considered to be an active medical device.
Any active medical device, whether used alone or in combination with other medical devices, to supply information for detecting, diagnosing, monitoring or treating physiological conditions, states of health, illnesses or congenital deformities is known as an active device for diagnosis.
Active devices intended for diagnosis are in Class IIa
if they are intended to image in vivo distribution of radiopharmaceuticals,
if they are intended to allow direct diagnosis or monitoring of vital physiological processes, unless they are specifically intended for monitoring of vital physiological parameters, where the nature of variations is such that it could result in immediate danger to the patient, for instance variations in cardiac performance, respiration, activity of CNS in which case they are in Class IIb.
Note the underlined wording above – in both the EU and the UK the software does not have to provide diagnosis or therapy itself to be so classified – it needs only to provide information used to make decisions.
Classifying the platform
The Platform itself may be a medical device or an accessory based on the level of functionality.
If the Platform itself can provide information used to make decisions, e.g. a PACS Platform that contains tools to measure features on radiological images or an EEG or ECG platform that provides tools itself to measure or interpret features of waveforms, then it needs to be classified accordingly, using the highest class applicable to the sorts of information it is providing.
If the Platform itself is used just to transmit or visualise information, then it could be an accessory – being careful that accessory in this context is a UK and EU MDR term, and defined differently in US regulations.
Under EU MDR, for instance, an accessory “assists the medical functionality” of a device, which fits the role of many platforms that support multiple MDSW apps through shared interfaces and infrastructure.
Certification and expansion
Ideally, the platform and its first MDSW app should undergo conformity assessment together:
If the Platform has Class I functionality, it can be self-certified by the manufacturer, provided it is accompanied by appropriate technical and clinical documentation.
However the first MDSW app if in Class IIa or above will need to be certified by a Notified Body (for CE marking) or a UK Approved Body (for UKCA marking). In this case, the Platform’s documentation must be included.
For CE marking under the EU MDR, two sets of information about accessories to an MDSW app (such as the Platform) are required:
A description of the accessories for a device, other devices and other products that are not devices, which are intended to be used in combination with it;
Technical specifications, such as features, dimensions and performance attributes, of the device and any variants/configurations and accessories that would typically appear in the product specification made available to the user, for example, in brochures, catalogues and similar publications
Additionally, instructions for use should provide information on the correct selection and use of accessories.
Once the Platform and the first MDSW app using the Platform have received a CE or UKCA mark, the next question is how to add further MDSW apps to the Platform.
If an MDSW app is from a different manufacturer to that of the Platform, contractual arrangements between the two parties – covering distribution, responsibilities, and liability – will need to be defined.
If the MDSW app is from the same manufacturer as the Platform, the goal is often to make the addition of new apps as quick and cost-effective as possible, ideally without repeating the full conformity assessment required for the first certification.
In such cases, manufacturers can rely on the EU MDR Annex IX provision stating that:
“The manufacturer in question shall inform the notified body which approved the quality management system of any plan for substantial changes to the quality management system, or the device range covered. The notified body shall assess the changes proposed, determine the need for additional audits and verify whether, after those changes, the quality management system still meets the requirements referred to in Section 2.2. It shall notify the manufacturer of its decision, which shall contain the conclusions of the assessment, and, where applicable, the conclusions of additional audits. The approval of any substantial change to the quality management system or the device range covered shall take the form of a supplement to the EU quality management system certificate.”
In practice, this means maintaining a transparent relationship with the Platform manufacturer’s Notified Body about plans to expand the device range. Robust design control and risk management procedures within the quality management system are essential to support these expansions while maintaining the safety, effectiveness, and cybersecurity of both the Platform and all associated MDSW apps.
So in this article I’ve presented the fundamental considerations. There are additional considerations, such as identification and traceability via UDI (unique device identifier) coding and version control ofMDSW apps vs the Platform itself, and the regulatory economic operator status of the Platform manufacturer (as a medical device manufacturer and/ or distributor, particularly if the Platform is being used to facilitate some kind of marketplace) to be covered in the future.
Hardian Health is a clinical digital consultancy focused on leveraging technology into healthcare markets through clinical strategy, scientific validation, regulation, health economics and intellectual property.