
Do I need to notify the MHRA about my clinical investigation?
Conducting premarket clinical investigations for Software as a Medical Device comes with regulatory risk, particularly when using uncertified products. Understanding when SaMD studies need MHRA Notice of No Objection (NoNo) is critical for avoiding delays. The MHRA’s updated guidance clarifies when notification is required, especially for studies involving patients or integration into live clinical workflows.
How do you know if your study needs a Notice of No Objection (NoNo)?
The MHRA has recently updated the notification guidelines to improve clarity for manufacturers and researchers. For software devices, conducting investigations involving patients or being used within a live clinical workflow are very likely to require a NoNo. There are specific situations in which notification may not be required, such as studies involving only retrospective data with no patient interaction and not informating patient management decisions. In situations where a device is being used for a medical purpose in an investigation (e.g. diagnosis of disease), an MHRA notification is likely to be required even if the results are not acted on in any way. Carefully examining the flowchart below, along with the accompanying guidance, will help manufacturers and researchers determine whether MHRA notification is required for a particular investigation.
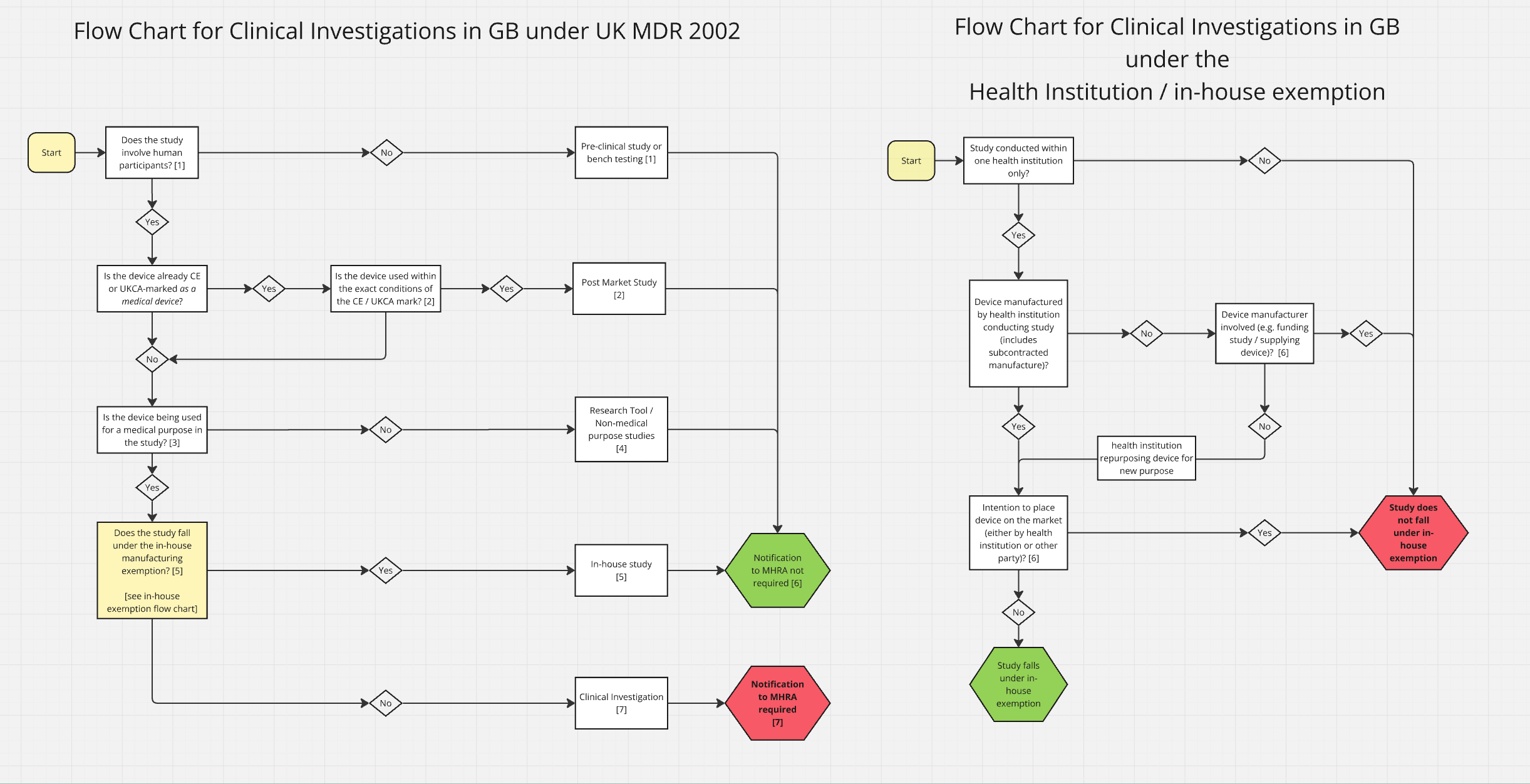
Flow chart for clinical investigations.
Once a device has achieved regulatory approval for use in the UK market, post-market studies will not require notification, provided that the device is still being used within its certified intended purpose. Where manufacturers are conducting post-market investigations to expand the intended purpose (e.g. using the device in a new patient population), this is likely to then also require MHRA notification.
How do I apply for an MHRA NoNo and what documents do I need to submit?
The process for submission has been streamlined in recent years and the MHRA NoNo submission is now made via the Integrated Research Application System (IRAS) at the same time as ethics and HRA approvals. There is a box to tick within the IRAS interface to indicate that MHRA review is also required. The MHRA notification must be submitted at least 60 days before the study start date. The current fee to the MHRA for most SaMD devices is £15,309, though a pilot fee waiver and payment easement processes are available for some SMEs and innovative technologies.

Clinical investigation for UKCA/CE UKNI/CE marking purposes
The MHRA has guidelines to support the compilation of documentation required for NoNo submission. A common misconception is that the same documents that are suitable for ethical review will also be suitable for MHRA NoNo submission. This is not the case. The MHRA review is largely focused on the risks posed by the unregulated medical device that will be used in the investigation. As a rule of thumb, the documentation required is generally 70-80% of the final Medical Device File (MDF). In particular the demonstration of thorough and complete risk management and tracing is required to satisfy the authorities.
This means every risk you identify must link directly to a specific design requirement, which in turn must link to a verification test proving it works. If you can’t "pull the thread" from a potential hazard all the way to a test result, your device isn't ready for MHRA NoNo submission. It's not just about having the right papers; it’s about proving the logic behind the safety.
Having this traceability ensures that the device is “safe enough” to be used for the purpose specified in the trial protocol (or Clinical Investigation Plan, in regulatory terms). There are several standards the MHRA will use to assess your documentation, and even as an early stage manufacturer, it is worth familiarising yourself with these:
ISO 13485:2016- Medical devices — Quality management systems — Requirements for regulatory purposes
ISO 14971:2019- Medical devices — Application of risk management to medical devices
IEC 62304:2006/AMD 1:2015- Medical device software — Software life cycle processes
What if I’m conducting my study outside the UK?
Each country has their own notification requirements for premarket clinical investigations of software as a medical device. For example, many European countries have similar notification requirements made to the relevant competent authority in that jurisdiction. Manufacturers should check local regulations in the country (or countries) in which the study is being conducted. In the US, the equivalent process is the investigational device exemption (IDE) submission conducted by Institutional Review Boards (IRB) at the clinical site(s) and/or the FDA. Some studies are exempt from IDE (meaning no IDE submission is required), while studies posing significant risk are subject to additional approval from the FDA and those with nonsignificant risk can follow an abbreviated IDE process via the IRB. Study sponsors are responsible for making an initial determination of risk level and presenting the case to the IRB.
Hardian has supported numerous successful MHRA NoNo submissions. Get in touch for help navigating this process and avoiding the common pitfalls.