
AI-Triage Devices: Essential Buyer's Guide for Healthcare
Buying AI-triage software isn't like purchasing regular tech. These tools are medical devices with strict regulatory requirements. Our guide (co-produced with Visiba Group) breaks down everything you need to know…
So, you’re considering AI-triage tools to help manage your overwhelmed clinics. The demos look impressive, the promises are compelling, and the vendor assures you everything is "fully compliant." Unfortunately, regulatory compliance isn't quite as straightforward as vendors would have you believe. It's illegal to market an AI-triage medical device without appropriate regulatory approval, and you're responsible for ensuring that what you're purchasing is legal to use.
We've encountered numerous healthcare organisations caught out by non-compliant AI tools. That's why we partnered with Visiba Group to create this buyer's guide. Consider it required reading before your next procurement decision.
AI-driven triage tools use artificial intelligence to assess patient symptoms and prioritise care. They guide patients to appropriate care levels and help clinicians make faster decisions. The key distinction lies in clinical influence.
Any AI-triage software that influences clinical decisions through summarising data, recommending urgency levels, or suggesting diagnoses must be regulated as a medical device.
Regulatory requirements and risk classifications
On to the nuts and bolts. Since 2021, the EU has operated under the Medical Device Regulation (MDR), which replaced older directives with stricter requirements. For EU/Northern Ireland markets, you need CE marking under EU MDR. For Great Britain, you need a UKCA marking or a valid CE mark (valid until June 2030).
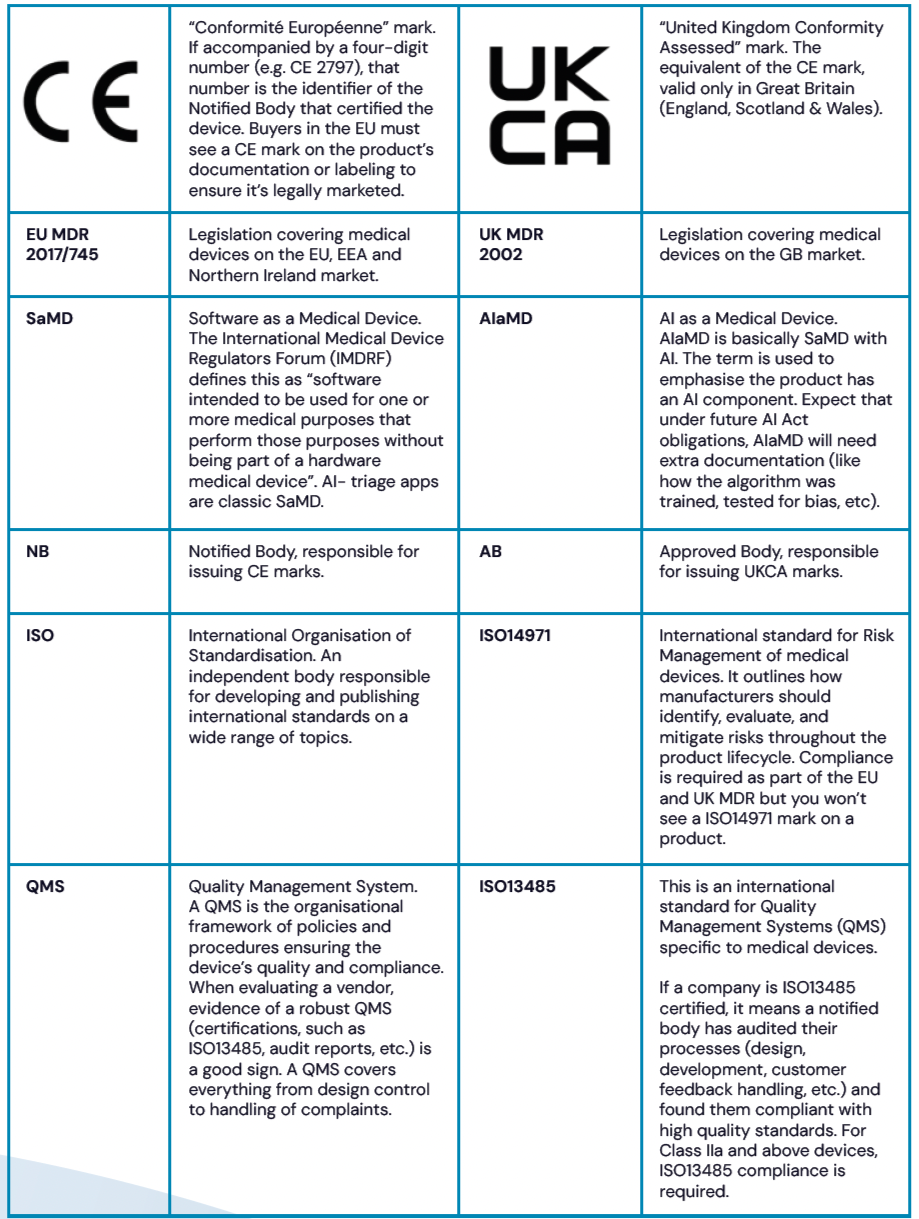
Table 1. Symbols, standards and acronyms
Many devices currently on the market were CE-marked under old regulations and have until 2028 to transition to EU MDR compliance, but only if they have a contract with a Notified Body. If your vendor hasn't started this transition, you may find yourself with an unusable system sooner than expected.
AI-triage tools should typically be, at least, Class IIa devices under EU MDR, requiring external verification of safety and performance. Be very wary of vendors claiming their tool is Class I under current regulations. Class IIb certification is required where errors could cause serious health deterioration.

Table 2. Medical device risk classification
Every medical device must have an Intended Use Statement defining exactly what the device does and in what context. Some tools may only be certified for adults or exclude certain symptoms. Always request complete documentation.
Local requirements and verification
Beyond basic compliance, vendors need to navigate additional local requirements that vary significantly by jurisdiction. In the UK, this includes DCB 0160 for clinical risk management and DTAC criteria, while different EU countries impose their own standards on top of MDR requirements. Fortunately, you can verify device registrations independently through the EUDAMED database for EU markets and MHRA PARD database for UK markets, or do it all in one place with our regulatory database HaRi, which helps cut through vendor claims.
Post-market surveillance adds another layer of ongoing compliance that many buyers overlook during procurement. Vendors must continuously monitor performance, collect user feedback, report adverse events, and produce periodic safety updates. The upcoming EU AI Act will layer additional requirements for medical AI devices by 2026, making vendor preparation increasingly important.
Essential compliance questions
Rather than taking vendor assurances at face value, six targeted questions will quickly reveal whether you're dealing with a compliant solution or a regulatory time bomb:
Is the product a certified medical device with proper CE/UKCA marking and appropriate risk classification?
Does the intended use statement match your specific requirements and patient population?
What clinical evidence supports the tool's performance in populations similar to yours?
How is patient data handled and what GDPR compliance measures are in place?
What post-market surveillance practices are implemented for ongoing safety monitoring?
How is the vendor preparing for upcoming EU AI Act requirements?
Start by confirming proper certification and risk classification, then ensure the intended use actually matches your specific requirements. Clinical evidence supporting performance in similar populations should be readily available, not promised for the future. Data handling deserves particular scrutiny given GDPR implications, so understand exactly how patient information flows through the system. Post-market surveillance practices reveal how seriously vendors take ongoing safety, particularly their processes for delivering updates and handling critical issues. Finally, gauge their preparation for EU AI Act requirements, which will separate forward-thinking vendors from those scrambling to catch up.
A genuinely compliant vendor will have ready answers, comprehensive documentation, and will actually appreciate your diligence because it demonstrates you understand the regulatory landscape. Remember that buying an AI-triage system means adopting a clinical tool that directly interacts with your patients and staff, so choose vendors who treat medical device regulations as fundamental rather than an inconvenient afterthought.
About the Authors
Dr Hugh Harvey, Dr Ankeet Tanna, Dr Felicity Lock, Dr Katherine Leung, Hannah Gibson, Angela Norton-Bilsby.
Based on our white paper co-produced with Visiba Group, experts in AI healthcare solutions and medical device compliance. For the complete guide, read here.
Hardian Health is a clinical digital consultancy focused on leveraging technology into healthcare markets through clinical strategy, scientific validation, regulation, health economics and intellectual property.